Gene therapy through different lenses
After decades of clinical research and numerous setbacks, the promise of gene therapy is now much more than a glimmering hope on the horizon.
Gene therapy is poised to revolutionize medicine as it presents the opportunity to remove or change the content of an individual’s genetic code in order to potentially treat or cure disease. [1] This is especially meaningful for those suffering from life-threatening monogenic diseases that appear at birth or begin to manifest within the first few months of life. And recent landmark product approvals around the globe, such as Kymriah, Yescarta, and Luxturna, represent the pioneers of this burgeoning new era in medicine.
Although there are only a handful of gene therapies presently available, many more are well on their way to hitting the market. Regulatory agencies have acknowledged a surge of cell and gene therapy products entering early development and only anticipate this trend to continue over the coming years. [2] In addition to the current 800 active cell-based or directly administered gene therapy Investigational New Drug (IND) applications on file, the FDA forecasts that by 2020 the agency will receive over 200 new IND applications annually.[2] They also predict that by 2025 up to 20 novel cell and gene therapy drugs will be approved annually.[2] The scene is changing, and changing quickly.
In order to address this massive influx of applications, the FDA is under pressure to hire more clinical reviewers and issue clinical guidance documents that will foster product development. These guidance documents should ideally serve a variety of stakeholders and help to navigate a new realm of medicine – no small task. Current and future gene therapy players are watching closely, as the guidelines and precedents being decided now will set the tone for years to come. Already there are numerous patient, payor, healthcare provider, and regulatory considerations that will need to be better understood and addressed to not only facilitate adoption, but ensure access. We take a look at gene therapy through these lenses below.
Patients
The appeal of gene therapy is evident for many patients – a potential one-time disease treatment or cure that provides them with the long-desired opportunity to return to good health. These innovative therapies can alleviate high treatment burdens, reduce compliance challenges, and encourage personalized medicine approaches. However, patients’ undeniable eagerness for this cutting edge technology is severely dampened by its blockbuster price tag.[4]
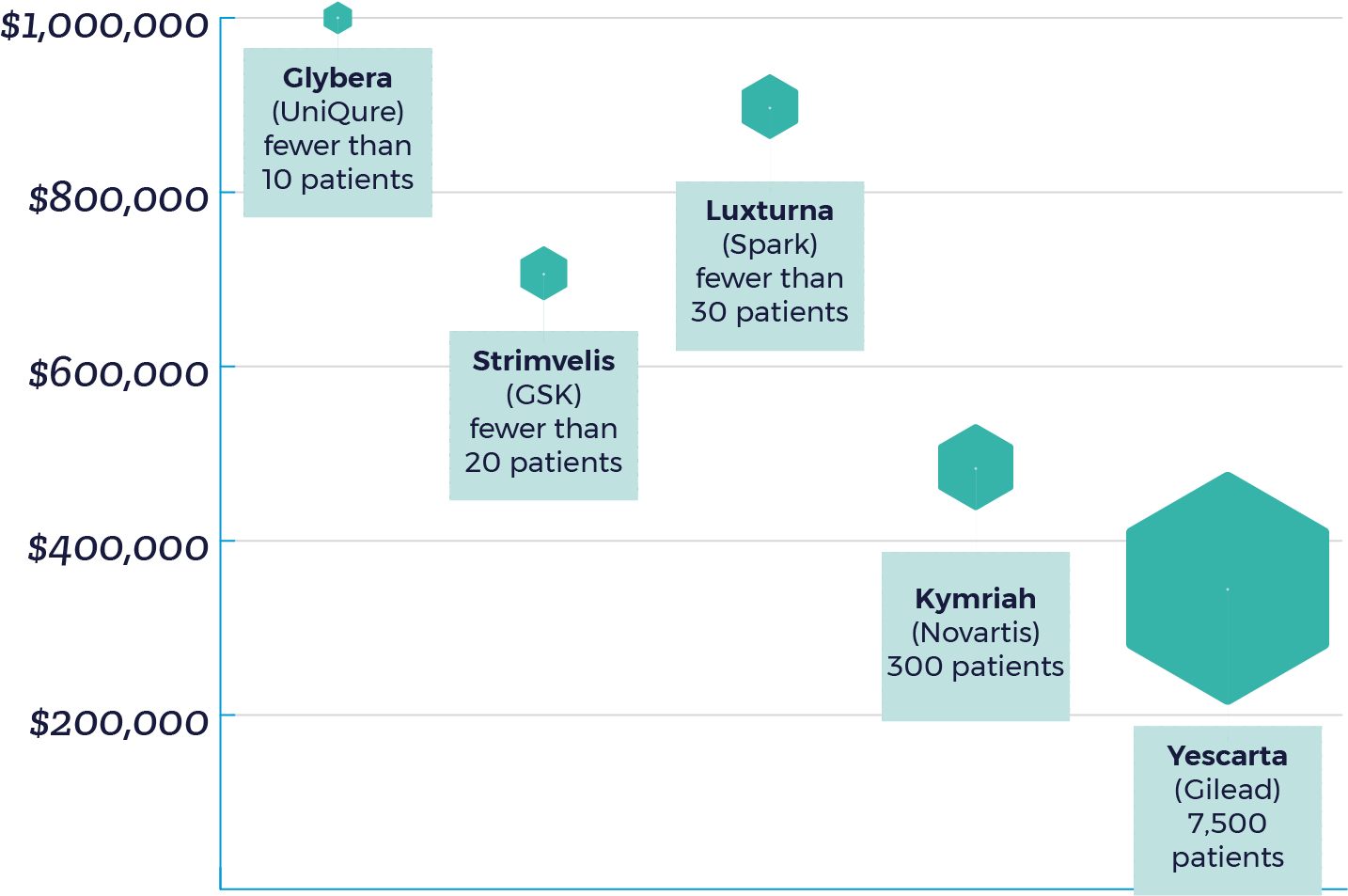
Without including the cost of hospitalization and medications for potential side effects, gene therapy prices have ranged from $373,000 to $1 million – with Glybera being withdrawn from the EU market due to limited usage given it’s $1 million price.[5] Due to high manufacturing costs and and limited patient populations, manufacturers have rationalized higher prices as a strategy to positive ROI. To put it simply: the fewer the patients, the higher the price.
It is important to highlight that the current market impact of approved therapies is limited as Kymriah and Yescarta are recommended only for cancer patients who have not responded to traditional treatments and Luxturna is indicated for a rare eye disorder. But as the innovative technology becomes more readily available for indications with larger eligible patient populations, such as hemophilia, affordability will ultimately determine the success of these life-changing agents. Cost is currently a fundamental treatment barrier for patients and must be addressed before additional players enter the market. Manufacturers will need to not only price their products appropriately, but also consider flexible payment methods to accelerate uptake.
There are many impactful patient advocacy organizations and disease-based foundations that have expanded their focus to promote gene therapy development and serve as a platform to build awareness.[3] For example, the co-founder of Sofia Sees Hope, Laura Manfre, testified before the FDA’s Cellular, Tissue, and Gene Therapies Advisory Committee (CTGTAC) in October 2017 as a representative of patients and families living with Leber congenital amaurosis (LCA), an inherited gene mutation that eventually leads to complete blindness. She used the platform to share moving patient stories involving the life-changing restoration of vision that could be made possible by gene therapy, and motivated the committee’s unanimous vote to recommend the FDA approval of Luxturna.[4] Her ability to appeal directly to decision-makers as the voice of those grappling with the disease points to the growing power patients can have on industry-changing treatment decisions.
Payors
With a new class of agents comes a new set of reimbursement challenges for manufacturers and payors that even the Institute for Clinical and Economic Review (ICER) is struggling to manage.[6] Although ICER has issued Final Evidence Reports for CAR-T therapies, Kymriah, Yescarta, and Luxturna, it acknowledges the difficulty in assessing the cost-effectiveness and value of these products at launch. And who can blame them? In the absence of long-term efficacy and safety data, it remains unknown whether these premium therapies offset total costs, especially if there is no existing treatment option. There is an evident need for more collaboration between stakeholders to generate robust clinical evidence and enable payors to make better informed coverage decisions.
“With many other potentially transformative therapies in the pipeline, stakeholders must collaborate now to develop payment and delivery systems that can ensure timely patient access, manage short-term affordability for expensive one-time treatments, and continue to reward the innovation that brings these new treatments to market.”
– Dan Ollendorf, PhD, ICER’s Former Chief Scientific Officer [10]
Reimbursement: Pricing options

Given that reimbursement frameworks are obsolete when it comes to these newer therapies, payors acknowledge the need for specific clinical evidence in order to assess product value and are seeking alternative payment models.[8] The lack of matured efficacy and safety data, which are generally guiding reimbursement principles, casts doubt on the durability of clinical benefit. Payors are posed with a significant financial risk if they provide the full payment upfront and a patient stops responding to one-time treatment. On the other hand, if insurers were to stagger payments over time in order to monitor treatment effect, a risk arises with patient healthcare plan portability. With either approach, the stakes remain high.
Value-based and outcomes-based contracting models are the predominant reimbursement approaches currently being undertaken in the US by gene therapy manufacturers to increase the rebate willingness of payors. Novartis, Gilead, and Spark have all tried different and creative variations to contracting approaches, but the need remains for a reimbursement model specifically tailored towards gene therapy products.
Thus, the FDA is currently working on six disease specific gene-therapy guidelines to provide manufacturers and payors with alternative data assessment recommendations, set the groundwork for future treatments and galvanize the development of methodical payment models.
HCPs
Despite the widely-recognized curative potential of gene therapy, healthcare providers remain apprehensive due to the lack of long-term safety data as well as the exorbitant treatment costs, which consequently impact shared treatment decision-making. Physicians aim to make well-informed decisions for their patients, but there are many caveats and nuances to consider when it comes to gene therapy.
If a patient is well-controlled on their current treatment regimen, should they seek gene therapy to potentially be disease-free? How does one communicate “potentially curative” without mismanaging patient expectations? What are the long-lasting treatment effects? What are the chances of off-target toxicity? And last, but certainly not least: is it worth the price?
These difficult and in some cases unanswerable questions suggest that initial gene therapy launches will only erode a small sector of patients, and that market uptake will only maximize when more long-term and real-world safety data becomes available. Thankfully, the FDA and EMA have required manufacturers to complete post-marketing studies in order to track patients and mitigate concerns over time. These patient registries will contribute to a growing body of evidence that healthcare providers can turn to when making important treatment decisions. Nonetheless, manufacturers will still need to invest in educational resources and support offerings to successfully familiarize healthcare providers along the way.
Regulatory agencies
With almost all new and emerging therapies comes the challenge of navigating through the complex regulatory landscape. Regulatory officials recognize the significant potential of gene therapy products and are working to refine the clinical development process via various broad and disease-specific gene therapy guidelines.[2] For example, in July 2018 the FDA issued a “Draft Guidance for Industry: Human Gene Therapy for Hemophilia,” to provide clinical trial design and preclinical considerations to support Hemophilia product development.[9]
This document highlights key considerations that will ultimately accelerate product development and approval in the future, and demonstrate the steadfast commitment of regulatory agencies to develop efficient pathways for gene therapy sponsors. However, given the sudden introduction of a new class of products, the recently published guidelines reveal considerations even the regulatory agencies are scrambling to identify and get in front of. Gene therapy manufacturers currently possess the unique opportunity to work in collaboration with regulatory agencies in order to establish fundamental requirements that will bring these novel treatments to market. Sponsors should engage in early and ongoing scientific dialogue with regulators to ensure alignment, mitigate potential setbacks in real-time, and fulfill requirements accordingly.
Spotlight on US regulatory policy
Gene therapy regulatory policies in the US are currently dynamic. The Regenerative Medicine Advanced Therapy (RMAT) designation and accelerated approval pathways provide manufacturers with the opportunity to expedite the product approval process using surrogate endpoints. However, given the FDA’s prediction that by 2025 up to 20 novel cell and gene therapy drugs will be approved annually, it will be important to better understand the common regulatory trajectory of gene therapy products.
Conclusion
As medicine enters a new era, and the use of gene therapy spreads across disease states, more pharmaceutical companies will want to enter this space. Look no further than Roche’s recent purchase of Spark Therapeutics for evidence of gene therapy’s allure and potential market power. This first wave of gene therapy products is attempting to pave the way for a new era of medicine and will lay the groundwork for gene therapy products yet to come.
As it stands, patients want a powerful cure that they can afford. Payors want reimbursement models designed specifically for the challenges posed by gene therapy. Healthcare providers want further confidence in such a potentially powerful treatment before they back it. And regulatory agencies are working hard to enable such a powerful scientific breakthrough to work as it was designed to do – change patients’ lives.
References
- https://www.asgct.org/education/more-resources/glossary
- https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm629493.htm
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4692103/
- https://www.patientsforaffordabledrugs.org/2017/10/18/taxpayers-and-us-patients-pay-twice-for-kites-new-car-t-drug/
- http://uniqure.com/GL_PR_Glybera%20withdrawal_FINAL_PDF.pdf
- https://www.managedcaremag.com/archives/2018/7/gene-therapy-must-sky-high-prices-come-down-price-right
- https://drugpricinglab.org/our-work/value-based-pricing-vs-outcomes-based-contracting/
- https://www.ncbi.nlm.nih.gov/pubmed/29144165
- https://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/UCM610801.pdf
- https://icer-review.org/announcements/car-t-final-report/